
Cranio-Fronto-Nasal Dysplasia


Abstract
Cranio-fronto-nasal dysplasia (CFND) is an X-linked dominant disorder, characterized by face and body asymmetry, midline face defects-cleft and/or high-arched palate, hypertelorism, bifid and broad nose, skeletal anomalies (limb anomalies, cranial synostosis) and ectodermal dysplasia (dental anomalies, grooved nails, wiry hair). Intellect is usually normal. We discuss the case of one patient, that presents with no family history, typical facial features (hypertelorism, bifid nose) skeletal anomalies (clubbed foot, coronal craniosynostosis), ectodermal dysplasia, brain anomalies (absent corpus callosum, internal hydrocephalus, parietal lobe atrophy, absent frontal bone) and intellectual disability. The particularity of this case is that intellectual disability was not expected, and when present, along with corpus callosum agenesis, usually points to a different diagnosis such as simple fronto-nasal dysplasia (FND).
Table of Contents:
1. Introduction
2. Case presentation and results
3. Discussions
1. Introduction
Cranio-fronto-nasal dysplasia (CFND) is an X-linked dominant disorder, characterized by face and body asymmetry, midline face defects-cleft or high-arched palate, hypertelorism, broad, bifid nose with a flat root. It associates skeletal anomalies (usually coronal synsostosis, which leads to brachycephaly and frontal bossing) and ectodermal dysplasia (dental anomalies, grooved, split nails, wiry hair) [1]. Corpus callosum agenesis has been reported in very few cases of CFND, although it seems not to interfere with intellectual development [2-4].
Paradoxically, even if it’s an X-linked disorder, females are much more severely affected whereas males are asymptomatic or present with a mild phenotype, frequently only displaying hypertelorism [1]. A possible explanation for this would be a metabolic interference between a mutant and a normal allele, leading to a more severe phenotype in heterozygous female patients than in hemizygous males [5, 6].
The responsible gene for this syndrome, EFNB1, has been mapped on the Xq13.1 region [1, 5]. The EFNB1 gene codes a protein (ephrine B1) that appears to be involved in the migration of neural crest cells, thus classifying this syndrome as a neurocrestopathy [4].
Reported mutations in this gene are missense, nonsense and frameshift mutations that result in premature termination codons (PTC) [2, 7].
2. Case presentation and results
The patient was first referred to the Genetics Department, as a newborn, for facial dysmorphism and limb anomalies. She was born at term, with an Apgar score of 8, from unrelated, young, healthy parents with a negative familial history. A fetal ultrasound during the 7th month of pregnancy detected a particular shape of the skull and a dilated left lateral ventricle which indicated c-section delivery.
She presented with frontal meningoencephalocele, thick, wiry hair, hypertelorism and right palpebral ptosis, flat nose, narrow and arched palate, asymmetric thorax, widely spaced and low set nipples, supernumerary nipples, and partial syndactyly of the 2nd and 3rd right toes. She had bilateral varus equinus and cast boots were applied right after birth. Height and weight were between normal parameters, but there was a slight microcephaly (Hc: 33 cm, -1,72 SD) which persisted through the following years. Also, in the following years there was a slight delay in her psychomotor development. At the age of 1 and 4 months old we raised the suspicion of coronal synostosis which was confirmed imagistically on a spiral CT scan. An MRI exam was also performed which diagnosed agenesis of the corpus callosum, internal hydrocephaly of the occipital and temporal horns of the lateral ventricles and slight parietal atrophy. She then underwent surgical treatment for coronal craniosynostosis at the age of 1 and 10 months old.
A molecular diagnosis by exon specific direct sequencing was made at the Otto-von- Guericke-Universität Magdeburg, in Germany. She was found to harbor a heterozygous frameshift mutation 28-231delCAAG/Y76fsX157 in exon 2 of the EFNB1 gene. This confirms the initial diagnosis of cranio-fronto-nasal dysplasia. The DNA of her parents was also tested with no mutation found. This is highly suggestive of a de-novo mutation. However, a germinal mosaicism in one of her parents cannot be excluded.
The last check-up in the Genetics Department was at the age of 8 years old (Fig. 1). Apart from the described features she had crowded teeth, more evidently split toe nails, and a moderate intellectual delay (struggled to keep up with school).
3. Discussions
The reason we chose to present this case is the particular intellectual disability associated with corpus callosum agenesis. While few cases have been reported in literature of CFND patients with corpus callosum agenesis, they did not associate any intellectual deficit [3, 4].
Moreover, frontal meningoencephalocele, agenesis of corpus callosum and intellectual deficit are more commonly associated with fronto-nasal dysplasia, an autosomal recessive syndrome caused by mutations of the ALX1 or ALX3 genes or an autosomal dominant syndrome with mutations on the ALX4 gene. This syndrome, however does not associate any of the ectodermal dysplasia features like the wiry, dry hair, dental anomalies or split nails [8-10].
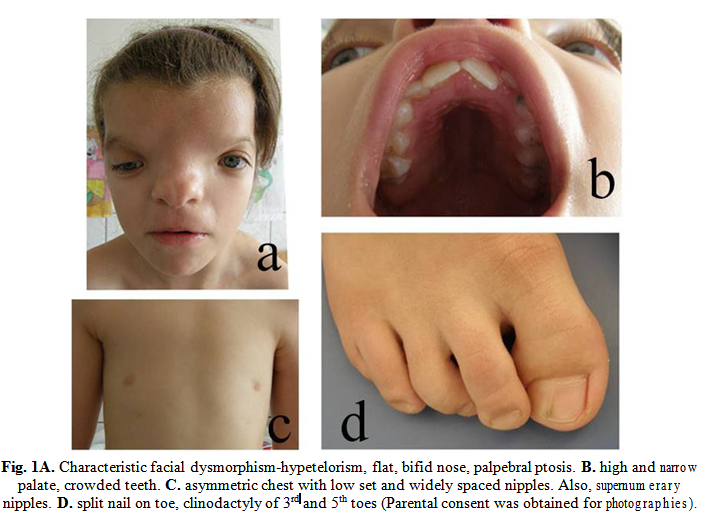
We eventually excluded the frontonasal dysplasia diagnosis after molecular testing that confirmed a frameshift mutation in the EFNB1 gene that is responsible for CFND.
Then why the intellectual disability in our case? Neurosurgical studies have shown that even an unilateral coronal synostosis was found to raise intracranial pressure (ICP) in some patients.
In infancy, a raised ICP due to craniosynostosis can have adverse effects on neurological development [11, 12, 13, 14]. Raised ICP in our case was excluded by an ophthalmic examination. There was no papillary edema which concluded that the patient’s ICP was normal.
However, other studies show that, most often in cases of unicoronal synostosis, but even in some cases of bicoronal synostosis, papilledema is not a consistent finding in children with raised ICP and even symptoms like blurry vision, emesis and headaches are late to appear [13, 15]. So we cannot conclude that intellectual disability in our case is syndromic or a consequence of raised intracranial pressure and late surgical correction of cranial synostosis.
Some studies recommend a constant invasive monitoring of intracranial pressure in children with craniosynostosis though acknowledging that this can be challenging to achieve in the pediatric population [11, 13]. These children should be closely monitored and raised ICP should be suspected in any case of non-syndromic craniosynostosis where even a slight delay of reaching development milestones is noted [14].
Given the above, we underlie the importance of continuous neurologic and neurosurgical monitoring of children with CFND. The syndrome in itself may not associate mental disability, but this can result as a secondary complication of the associated craniosynostosis. It is important to remember that classical findings of raised ICP are not always present in children with craniosynostosis and evaluating the patient further is often necessary.
The Authors:
AANICĂI Ruxandra [1]
WIEACKER Peter [2]
RUSU Cristina [3] [4]
[1] “Cuza-Vodă” Clinical Hospital of Obstetrics and Gynaecology, Department of Genetics, Iaşi, (ROMANIA)
[2] “Otto-von-Guericke” Universität Magdeburg, Institut für Humangenetik,(GERMANY)
[3] “Sf. Maria” Paediatric Emergency Hospital, Regional Medical Genetics Centre, Iaşi, (ROMANIA)
[4] “Grigore T. Popa” University of Medicine and Pharmacy, Medical Genetics Department, Iaşi, (ROMANIA)
Contributo selezionato da Filodiritto tra quelli pubblicati nei Proceedings “5th Medical Genetics Congress with International Participation - 2018”
Per acquistare i Proceedings clicca qui.
Contribution selected by Filodiritto among those published in the Proceedings “5th Medical Genetics Congress with International Participation - 2018”
To buy the Proceedings click here.
